If you have ever heard the phrase “For the sake of argument..”, you’ve heard a hypothesis. A hypothesis is a proposition about the world that forms the starting point for an investigation. A hypothesis doesn’t have to be true, and oftentimes it can be helpful to start from a hypothesis that is probably not true.
For example, let’s start with the hypothesis that vanilla is the favorite ice cream flavor of all Tufts students. This may not be true, and in fact it probably isn’t true. But it provides a useful basis for investigation: if that were true, then every time I asked a student on campus their favorite ice cream flavor, they would reply “vanilla”. The moment I received another answer, such as “chocolate” or “dill pickle”, then my hypothesis would be rejected. By rejecting my hypothesis, I’ve learned about the ice cream preferences of the students.
A hypothesis test, then, is a test that determines whether that hypothesis should be kept or rejected. The main goal of testing a hypothesis is not to prove it is true, but to rule it out as a possibility. Hypothesis tests are principally used to establish either a) a difference between variable values between groups; or b) a relationship between two or more variables.
The Process
There are four basic components to a hypothesis test:
Stating the hypothesis
Collecting the data
Conducting a test
Interpreting the p-value
We’ll go through each one to get a clearer idea of what each one means. If you’re a little rusty with stats, it’s worth reading the section below as it provides a good basis for understanding the use of hypothesis testing. If you feel like you’re pretty familiar with stats and how hypothesis testing works, you can skip to the Hypothesis testing in R heading.
State the hypothesis
It’s really important to start with a clear statement of the hypothesis with clear criteria for rejection. In the ice cream example above, the statement “Vanilla is the favorite ice cream flavor of all Tufts students” has a clear way to reject it: find someone who prefers another flavor.
While we could formulate hypotheses in many ways, a helpful place to start is a null hypothesis: this is the idea that there is no difference or relationship among the variables, and that any apparent difference/relationship in the sample data is the result of chance. For example, if we were interested in whether trees in one city were larger on average than trees in another, the null hypothesis would be “there is no difference in the average height of trees in the two cities”.
This idea of chance is really important, because when we don’t know what process generated the pattern, it is always possible that a null hypothesis is actually correct. We can think of a null hypothesis test, then, as a statement of how likely it is that a null hypothesis is true. So before we move on, we also need to establish the criteria we would use to reject the null hypothesis. A widely held convention is to reject the null hypothesis if there is less than a 5% chance that it is true (see below).
We may also want to evaluate an alternative hypothesis: this is an expectation about the nature of difference or relationship. It is usually expressed in terms of an direction (e.g., less than, greater than)1. For example, an alternative hypothesis in our tree height scenario is “Trees in Charlotte, NC are larger than those in Charlestown, SC”. It might also just be the opposite of the null hypothesis without stating a direction2: “Trees in Charlotte, NC are differently sized than those in Charlestown, SC”
Collect the data
The next step is to collect the data, usually by sampling variables within a population. In data science, the collection process is often involves gathering and repurposing of existing datasets. As such, we don’t necessarily get to control how data is collected, and must evaluate the quality of the data post hoc. The first thing to consider is, in the context of your hypothesis, what will be the dependent (or response) variables, and what will be the independent variables (if any). For example, if your hypothesis is that the average size of wildfires in Colorado have not changed between the years 2000 and 2020, you would need a response variable (wildfire sizes in Colorado) and one independent variable (the year of the fire).
Perform a test
Next, we perform our test using the data we collected. You’ve likely (hopefully) heard of some of the common statistical tests: chi-squared test, t-test, Pearson correlation test, etc. The particular test being used will be determined by the kind of hypothesis (e.g., looking for a difference, looking for a relationship), the kind of data (e.g., nominal, ordinal, interval, ratio), and how the data were collected (independent samples, repeated measures, etc). We’ll look at the process of choosing a test in the next section.
Each test will also come with a set of assumptions about the composition of the data. For example, a common assumption is that the error/uncertainty is normally distributed, which is based on the idea that there isn’t some other process biasing the distribution.
Interpret the p-value
Once we have calculated the test statistic, we can determine the p-value, which is the that probability that the difference or relationship observed in the sample could arise under a given hypothesis. We then determine whether this probability is enough to retain the hypothesis or reject it.
In the case of testing a null hypothesis, a p-value of 0.05 would indicate that the observed difference or relationship has a 5% probability of arising by chance; in other words, it is highly unlikely. This is a commonly used standard in many scientific disciplines.
Keep in mind that a high p-value or a low p-value is not an indicator of the relative strength of any hypothesis. Instead, it is an indicator of the evidence against the null hypothesis.
Hypothesis testing in R
There are a number of functions in R that provide access to hypothesis tests, and even more that can be accessed in specialized statistics packages. Each, but they usually follow something like the following format:
test.name(variable1, variable2, arguments)
The variables in this example are typically vectors of values, which might come from a column in tibble or dataframe. The vectorized operation of these functions makes conducting hypothesis tests in R a pretty straightforward process.
However, there is a catch: as long as you give it the correct kinds of arguments, R will give you some kind of result, even if it is not appropriate for your data or your hypothesis. Therefore, it is up to you to be skeptical, carefully determine whether the appropriate test has been selected, and interpret the results accordingly.
A motivating example
To get a sense of how this all works, let’s look at the penguins dataset from the palmerpenguins package. Let’s say that a study showed that Chinstrap and Gentoo penguins at Cape Adare in Antarctica have the same average body mass. We want to know whether the same thing is true for the Palmer Islands penguins population.
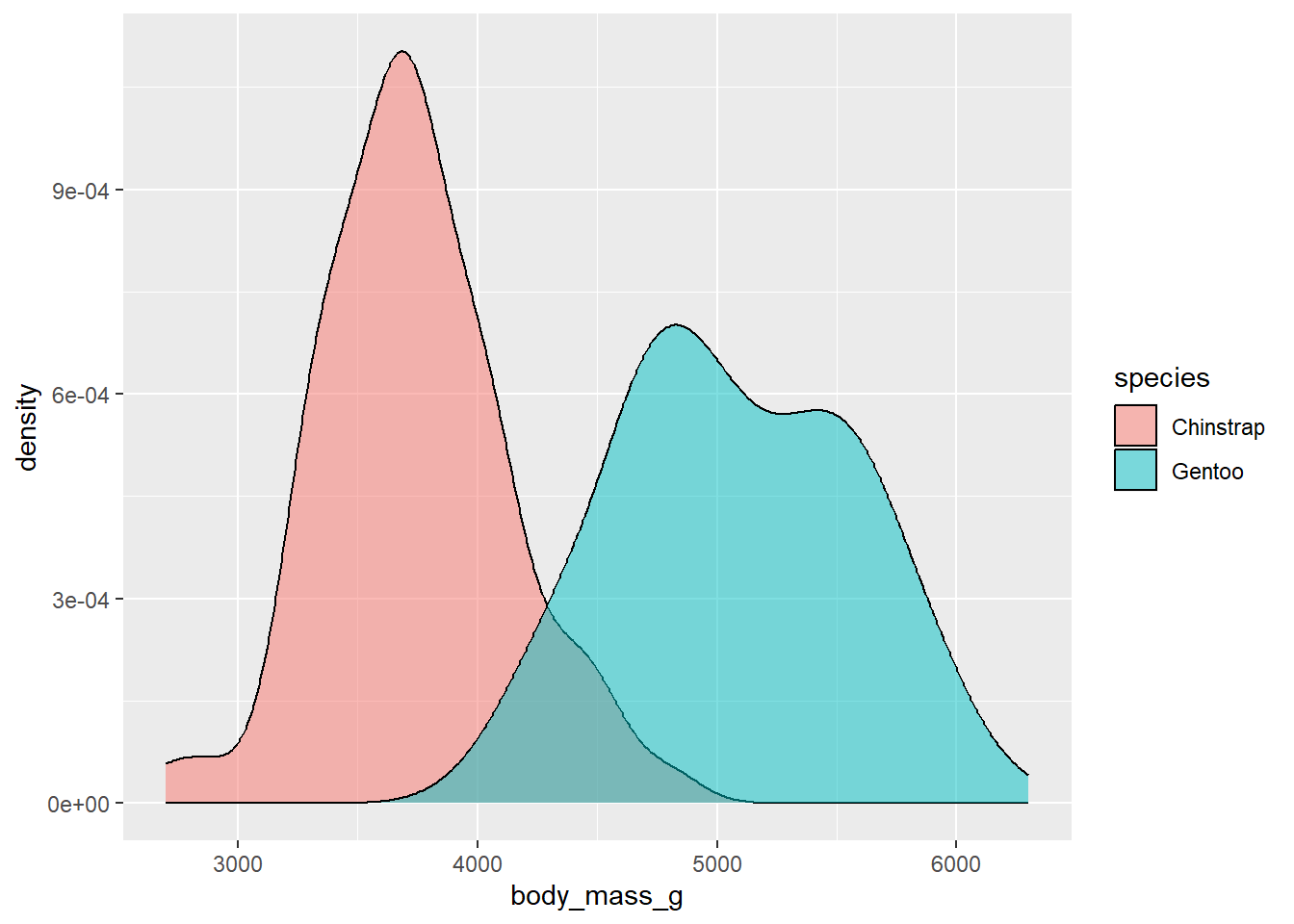
First, let’s look at the body mass distributions of these two penguin species in our dataset:
library(palmerpenguins)#create a dataset of just Adelie and Gentoo penguins, and just the species and body mass columnsgenChin <- penguins %>%filter(species =="Chinstrap"| species=="Gentoo") %>%select(species, body_mass_g) %>%drop_na()(#plot the two speciesggplot(genChin,aes(x=body_mass_g,fill=species))+geom_density(alpha=0.50))
Here you can see that body masses among the samples of penguins vary between 2500 an 6500 g3. Each species’ values also appear to follow a roughly normal distribution around a central (mean) value. We can get the average body weight of these samples by using the mean function:
So the average body mass for Chinstrap penguins in our sample is around 3733 grams, while the average for Gentoo penguins is around 5076. Just looking at these values would suggest there is a difference between the two populations. However, it is also possible that this is simply an outcome of our sampling. What if their average body mass were the same, and by random chance we just happened to get larger Gentoo penguins and/or smaller Chinstrap penguins?
We can reformulate this question as our null hypothesis: the average body mass of the population of Palmer Islands Chinstrap penguins is no different than that of the Gentoo penguins. If that were the case, then what is the probability that our samples would have a difference of 1376 g?
Next, we collect our data: in this case, it’s the body masses of the Chinstrap and Gentoo penguins, which we already assembled as a table above:
Different kinds of hypotheses and data require different tests. Knowing which test is appropriate for which situation takes a lot of practice, and even among researchers who regularly use of statistics it is common to lo. Even looking up the right test can be tricky if you don’t know exactly what kinds of things you need the test to handle.
The test we want to conduct is looking for differences in a continuous/ratio variable (body mass) with two independent samples (Chinstrap and Gentoo penguins). A common test used for this kind of analysis is the independent two-sample t-test. This calculates whether the difference in the means of two samples might arise from sampling two larger populations with the same mean.
The t.test function allows us to run this test in R. This function takes, as minimum, two arguments: a data argument (a tibble or dataframe), and formula argument that gives two vectors connected by a tilde (~). R interprets the formula x ~ y as “compare variable x by variable y”, where y refers the groups/samples being compared. Here’s what it looks like:
t.test(data = genChin, body_mass_g ~ species)
Welch Two Sample t-test
data: body_mass_g by species
t = -20.628, df = 170.4, p-value < 2.2e-16
alternative hypothesis: true difference in means between group Chinstrap and group Gentoo is not equal to 0
95 percent confidence interval:
-1471.440 -1214.416
sample estimates:
mean in group Chinstrap mean in group Gentoo
3733.088 5076.016
This is a pretty typical output from an R statistical test: it tells us the name of the test, what data is being assessed, and then a set of values derived from the test. How do we interpret these numbers?
t This is the test statistic for the t-test. Values of t that vary from zero suggest a difference between the group means
df = This is the degrees of freedom, which is based on the number of individuals in each sample. This is used in the calculation of the p-value.
p-value This is the probability that the averages of our sampled penguin species arose by sampling larger populations with no difference between them.
alternative hypothesis Since we didn’t specify an alternative hypothesis, the default is the direct alternative to the null hypothesis: that there is a difference in the average body masses of the two penguin species.
confidence interval This is a range of values within which the true population mean is expected to fall. Since we haven’t measured the population at large, this gives us the
mean in group This is the mean of each sample (see above)
One thing to note here is that the method here is the Welch method. This method is used when the samples have unequal variances, and R uses this as the default. We’ll briefly touch on why this matters further down.
Looking at our p-value, we see 2.2e-16. This is a scientific notional rendering of 2.2 x 10-16, or 0.00000000000000022. In other words, a number very close to zero. This is about as low a number as R will register for a p-value, and is certainly lower than the 0.05 criteria for rejection. So our interpretation is that there is not enough evidence to retain the null hypothesis.
Keeping track of test assumptions
Each hypothesis test will come with it’s own set of assumptions about the condition of the data. If these assumptions are not met, then any result from these tests might be considered suspect.
The main assumptions of the t-test are as follows:
The data are independent (in this case, the body mass of one penguin doesn’t affect that of another)
The sample was collected randomly from the population
Data are normally distributed (e.g., not positively or negatively skewed)
Some of these will be obvious based on the type of data: for example, we can say confidently that one penguin’s body mass is not directly influencing that of other penguins. Others we will not have direct control over as data scientists. For example, we can’t say for certain whether the sample of penguins would qualify as completely random, and so this has to remain an assumption.
Still others we can test whether they meet the criteria of the test by using the data itself. In this case, we can assess whether the data are normally distributed. For example, the Shapiro-Wilk test allows us to assess normality in a vector of values. It is a null hypothesis test itself, where the null hypothesis is that the data are drawn from a normal (non-skewed) distribution.
We can use the shapiro.test function to access this test. First, we’ll look at distributions for Gentoo penguins:
shapiro.test(gentoo$body_mass_g)
Shapiro-Wilk normality test
data: gentoo$body_mass_g
W = 0.98593, p-value = 0.2336
Now for Chinstrap penguins:
shapiro.test(chinstrap$body_mass_g)
Shapiro-Wilk normality test
data: chinstrap$body_mass_g
W = 0.98449, p-value = 0.5605
Since both of these values are greater than 0.05, we cannot reject the null hypothesis that these were drawn from normally distributed data. So on this front, our analysis stands.
Finally, remember above that R default to using the Welch method in a two-sample t-test. In a standard t-test, another assumption is that the variance between the two samples is equal. Over the years several methods have been developed that allow a hypothesis test when certain assumptions cannot be met, and the Welch method is one of those. But was it necessary for our analysis?
The F-test provides us with a way to test for equal variance. The F-test is another form of null hypothesis test, but the hypothesis in this case is that there is no difference in the variance (how far the values extend around the mean) in body mass in the two groups.
We can access this in R using the var.test function:
var.test(body_mass_g ~ species, data=genChin)
F test to compare two variances
data: body_mass_g by species
F = 0.58124, num df = 67, denom df = 122, p-value = 0.01559
alternative hypothesis: true ratio of variances is not equal to 1
95 percent confidence interval:
0.3852832 0.9004223
sample estimates:
ratio of variances
0.5812443
In this case, our p-value is under 0.05, so we can say that there is no evidence that our samples have the same variance. That means that using the Welch method was the right call for our data.
tl;dr
Hypothesis tests are a way to establish whether a difference or relationship within a dataset is likely to have arisen. The process of conducting a test involves multiple steps. Here, we used the two-sample t-test as an example of how to go about conducting a test with a particular kind of data and hypothesis. Each situation will require a determination of the kind of test to be used. In the next section, we’ll look at how to incorporate this into the process.
This is called a one-sided alternative hypothesis.↩︎
This is called a two-sided alternative hypothesis.↩︎
Note here that the density plot appears to continue beyond these values. The edges of the density plot are set by the limits of the data itself. If you wanted to modify this to show the entire distribution, you could set these manually using xlim↩︎